
GeMoMa to merge annotations#
Author:
📝 Prerequisites#
📁 Input files
Target genome assembly (
fastaformat) - we will use B73.v5 genomeab initio predictions (
.gff3format) - we will use Helixer outputHomology predictions (
.gff3format) - Sorghum and B97 annotations
💻 Software
GeMoMa (version ≥ 1.9 recommended)
Dependencies:
java >1.8,blastormmseqs
🧬 GeMoMa overview#
Gene Model Mapper (GeMoMa) is a homology-based gene prediction tool that transfers protein-coding gene models from one or more reference genomes to a target genome. It can incorporate RNA-seq evidence, filter predictions using customizable criteria, and merge multiple annotations—including ab initio and transcriptome-based predictions—into a unified gene set.
🔍 Use case: merging annotations#
Helixer is a deep learning-based predictor that provides accurate gene predictions from genome sequence alone. However, it does not predict alternative isoforms and tends to collapse all transcript variants into a single flattened gene model, especially in regions with overlapping exons or alternative splicing.
Homology predictions can be used to recover these missing isoforms and provide additional context for gene structure. GeMoMa excels at this task by leveraging evolutionary conservation across related species, allowing it to transfer annotations from well-annotated genomes to the target genome.
By merging Helixer predictions with GeMoMa:
🧠 You retain Helixer’s high confidence gene boundaries and structure for primary transcripts.
🔁 You recover missing isoforms and splice variants that Helixer collapses.
🔍 You gain orthology-supported annotation refinement using multiple maize lines and related species like Sorghum bicolor.
🧬 You generate a more complete and biologically realistic gene set, even without long-read or isoform-resolving RNA-seq data.
This hybrid strategy takes advantage of both machine learning accuracy and evolutionary conservation to compensate for the limitations of any single method.
🗂️ Input preparation#
In this example, for the B73.v5 genome, we will merge ab initio predictions obtained from Helixer with the homology predictions of its close relatives (Maize inbred B97 and Sorghum).
Download target genome:
wget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0.fa.gz
Download Helixer predictions:
wget
Download homology predictions (genome and gff3 files):
wget https://download.maizegdb.org/Zm-B97-REFERENCE-NAM-1.0/Zm-B97-REFERENCE-NAM-1.0.fa.gz
wget https://download.maizegdb.org/Zm-B97-REFERENCE-NAM-1.0/Zm-B97-REFERENCE-NAM-1.0_Zm00018ab.1.gff3.gz
wget https://ftp.sorghumbase.org/release-9/fasta/sorghum_bicolor/dna/Sorghum_bicolor.Sorghum_bicolor_NCBIv3.dna.toplevel.fa.gz
wget https://ftp.sorghumbase.org/release-9/gff3/sorghum_bicolor/Sorghum_bicolor.Sorghum_bicolor_NCBIv3.gff3.gz
Unzip the downloaded files:
gunzip Zm-B73-REFERENCE-NAM-5.0.fa.gz
gunzip Zm-B97-REFERENCE-NAM-1.0_Zm00018ab.1.gff3.gz
gunzip Sorghum_bicolor.Sorghum_bicolor_NCBIv3.dna.toplevel.fa.gz
gunzip Sorghum_bicolor.Sorghum_bicolor_NCBIv3.gff3.gz
🚀 Setup GeMoMa#
GeMoMa is executed using a Java-based CLI pipeline. Here we will run GeMoMa with the target genome, Helixer predictions, and homology-based annotations from related species. We will generate a merged annotation file in GFF3 format.
We will install GeMoMa using BioConda:
module --force purge
module load conda
conda create -n gemoma # Press 'y' when prompted
conda activate gemoma
conda install -c bioconda gemoma
After installation, the GeMoMa JAR file is located under the environment’s shared directory. You can set the path like this:
gemoma_jar="${CONDA_ENVS_PATH}/share/gemoma-1.9-0/GeMoMa-1.9.jar"
🛠️ Run GeMoMa#
#!/bin/bash
# activate conda environment
module --force purge
module load conda
conda activate gemoma
# Set GeMoMa JAR path
gemoma_jar="${CONDA_ENVS_PATH}/share/gemoma-1.9-0/GeMoMa-1.9.jar"
# Function to check file existence and resolve full path
check_file() {
if [[ ! -f "$1" ]]; then
echo "ERROR: File not found: $1" >&2
exit 1
fi
realpath "$1"
}
# Check and assign input files
genome=$(check_file Zm-B73-REFERENCE-NAM-5.0.fa)
sorghumFa=$(check_file Sorghum_bicolor.Sorghum_bicolor_NCBIv3.dna.toplevel.fa)
sorghumGFF=$(check_file Sorghum_bicolor.Sorghum_bicolor_NCBIv3.gff3)
maize1=$(check_file Zm-B97-REFERENCE-NAM-1.0.fa)
maize1GFF3=$(check_file Zm-B97-REFERENCE-NAM-1.0_Zm00018ab.1.gff3)
helixer=$(check_file Zm-B73-HELIXER-NAM-1.0.gff3)
# Other config
name="B73_v5_GeMoMa" # Prefix for gene names
cpus=${SLURM_CPUS_ON_NODE:-8} # Default to 8 CPUs if not running on SLURM
TMPDIR=${TMPDIR:-/tmp} # Use system temp directory if not set
dir="gemoma_output" # Output directory
# Run GeMoMa
java -Xms100g -Xmx200g -Djava.io.tmpdir=$TMPDIR -jar ${gemoma_jar} CLI GeMoMaPipeline \
t=$genome \
s=own i=sb a=$sorghumGFF g=$sorghumFa \
s=own i=zm a=$maize1GFF3 g=$maize1 \
ID=helixer e=$helixer \
AnnotationFinalizer.u=NO \
AnnotationFinalizer.r=SIMPLE AnnotationFinalizer.p=$name \
AnnotationFinalizer.n=true \
sc=false o=false p=true pc=true pgr=false \
threads=$cpus outdir=$dir &> ${dir}/gemoma.log
Save this script as run_gemoma.sh and run it in a terminal with the command:
chmod +x run_gemoma.sh
./run_gemoma.sh
The options used in the command above are:
Option |
Explanation |
|---|---|
|
Target genome (FASTA) to annotate. |
|
Reference species data will be extracted by GeMoMa from annotation + genome. |
|
Identifier for reference species (Sorghum bicolor). |
|
GFF3 gene annotation for Sorghum bicolor. |
|
FASTA genome for Sorghum bicolor. |
|
Identifier for second reference species (e.g., maize B97). |
|
GFF3 gene annotation for maize B97. |
|
FASTA genome for maize B97. |
|
Unique label for Helixer-based annotation source. |
|
External GFF3 annotation to be merged (Helixer output). |
|
Disable UTR prediction (no RNA evidence available). |
|
Rename gene/transcript IDs using a simple prefix-based system. |
|
Prefix used in renaming genes and transcripts. |
|
Add new names as |
|
Disable synteny check. |
|
Do not write individual predictions per reference species. |
|
Output predicted proteins as FASTA. |
|
Output predicted CDSs as FASTA. |
|
Do not output predicted genomic regions. |
|
Number of compute threads to use. |
|
Output directory for all GeMoMa results. |
📈 Interpreting results#
The GeMoMa output will be stored in the specified gemoma_output directory. The main results include:
final_annotation.gff: Merged GFF3 annotation file containing gene models from Helixer and homology predictions.predicted_proteins.fasta: FASTA file with predicted protein sequences.predicted_cds.fasta: FASTA file with predicted coding sequences (CDS).
If you examine the log file (gemoma.log), you will see some important stats, including any warnings or errors encountered during the run.
You can also check the number of genes and transcripts in the final annotation:
grep -v '^#' gemoma_output/final_annotation.gff | cut -f3 | sort | uniq -c
Which should give you the counts for all features.
246097 CDS
58847 gene
60137 mRNA
You can clean up the gff file using the genometools utility:
ml --force purge
ml genometools
ml agat
gff3_file=gemoma_output/final_annotation.gff
gt gff3 \
-sort \
-tidy \
-setsource "gemoma" \
-force \
-o B73-gemoma_gt.gff3 \
$gff3_file
agat_convert_sp_gxf2gxf.pl \
-g B73-gemoma_gt.gff3 \
-o B73-gemoma_v1.0.gff3
gffread B73-gemoma_v1.0.gff3 \
-g Zm-B73-REFERENCE-NAM-5.0.fa \
-x B73-gemoma_v1.0.cds.fasta \
-y B73-gemoma_v1.0.pep.fasta
🔎 Quality assessment#
A. BUSCO profiling#
Run BUSCO to assess the completeness of the merged annotation:
ml --force purge
ml busco
busco \
-i B73-gemoma_v1.0.pep.fasta \
-c $SLURM_CPUS_ON_NODE \
-o B73-gemoma_v1.0_busco \
-m prot \
-l poales_odb10
We can compare the results with the previously generated Helixer and B73.v5 annotations and whole genome assembly BUSCO results.

Figure 1: BUSCO results for Helixer, B73.v5 (MaizeGDB) and merged (GeMoMa) annotations.
B. Comparing annotations#
Compare Helixer original predictions with GeMoMa:
conda activate mikado
mikado compare \
--protein-coding \
-r Zm-B73-HELIXER-NAM-1.0.gff3 \
-p B73-gemoma_v1.0.gff3 \
-o ref_NAM.B73v5-vs-prediction_HELIXERv1_compared \
--log compare.log
Compare B73.v5 (MaizeGDB) predictions with GeMoMa:
mikado compare \
--protein-coding \
-r Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3 \
-p B73-gemoma_v1.0.gff3 \
-o ref_NAM.B73v5-vs-prediction_HELIXERv1_compared \
--log compare.log
D. Functional annotation#
The Eukaryotic Non-Model Transcriptome Annotation Pipeline (EnTAP) can be used for functional annotation of the predicted genes.
ml purge
ml anaconda
conda activate entap
cds="B73-gemoma_v1.0.cds.fasta"
EnTAP \
--runP \
-i ${cds} \
-d ${RCAC_SCRATCH}/entap_db/bin/ncbi_refseq_plants.dmnd \
-d ${RCAC_SCRATCH}/entap_db/bin/uniprot_sprot.dmnd \
-t ${SLURM_CPUS_ON_NODE} \
--ini ${RCAC_SCRATCH}/entap_db/entap_config.ini
E. Summary Statistics#
You can summarize the results using the agat utility:
ml biocontainers
ml agat
agat_sp_statistics.pl \
-g B73-gemoma_v1.0.gff3 \
-o B73-gemoma_v1.0.stats
F. OMArk proteome assesment#
OMArk can be used to assess the quality of the predicted proteome:
ml --force purge
ml seqtk
omark_sif="${RCAC_SCRATCH}/omark/omark_0.3.0--pyh7cba7a3_0.sif"
peptide="B73-gemoma_v1.0.pep.fasta"
database="${RCAC_SCRATCH}/omark/LUCA.h5"
grep ">" ${peptide} |\
sed -e 's/gene=/\t/g' -e 's/>//g' |\
sort -uk2,2 |\
cut -f 1 > primary.ids
seqtk subseq ${peptide} primary.ids > ${peptide%.*}.primary.pep.fasta
apptainer exec ${omark_sif} omamer search \
--db ${database} \
--query ${peptide%.*}.primary.pep.fasta \
--out ${peptide%.*}.omamer \
--nthreads ${SLURM_CPUS_ON_NODE}
G. CDS assesments#
cds="B73-gemoma_v1.0.cds.fasta"
bioawk -c fastx 'BEGIN{OFS="\t"}{
print $name,length($seq),gc($seq),substr($seq,0,3),reverse(substr(reverse($seq),0,3))
}' ${cds} > ${cds%.*}.info
The plots were generated using the following R script: cds_assesment.R
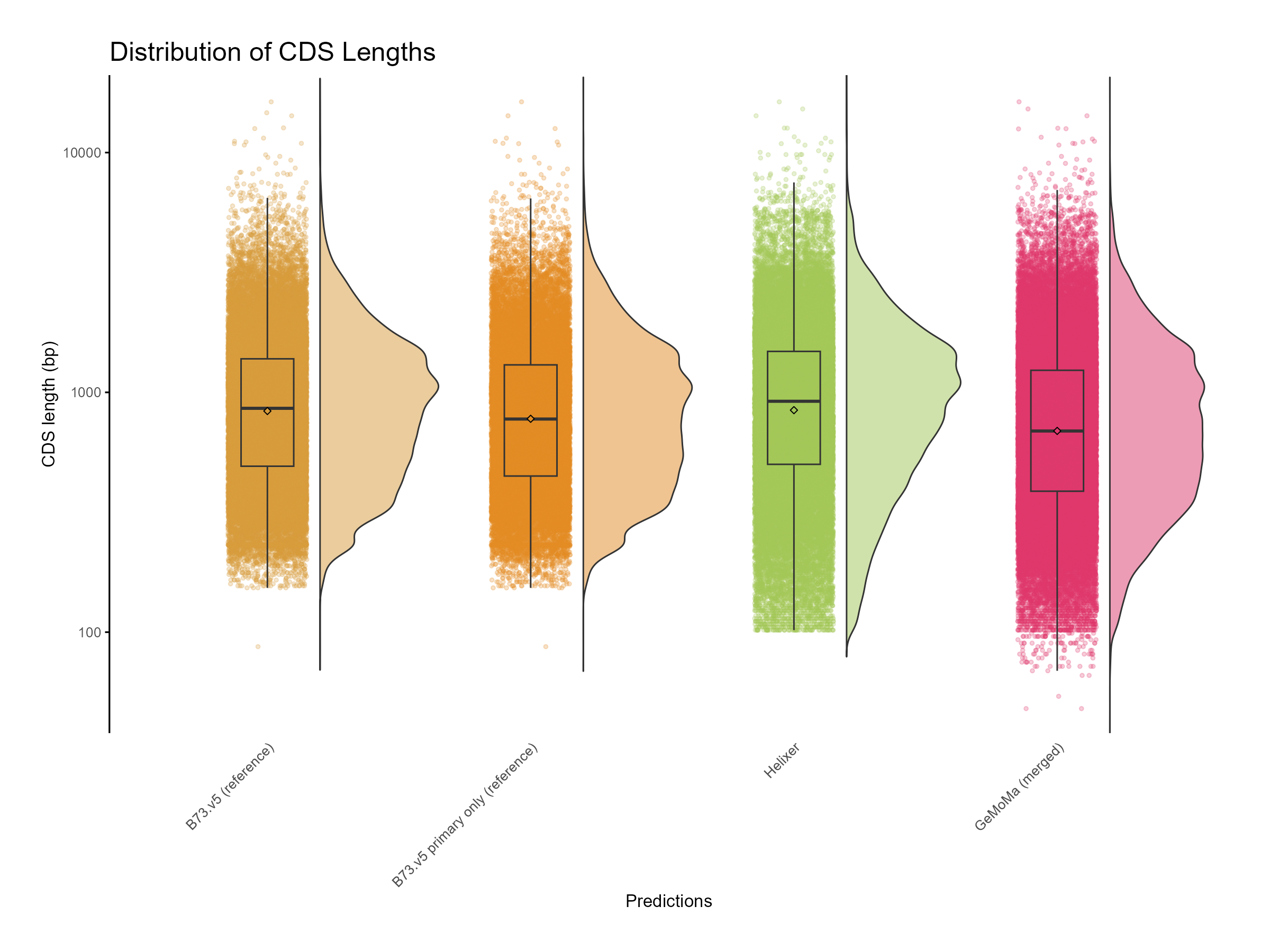
Figure 8: Distribution of CDS length for GeMoMa, Helixer and NAM.v5 predictions.

Figure 9: Distribution of GC content for GeMoMa, Helixer and NAM.v5 predictions.
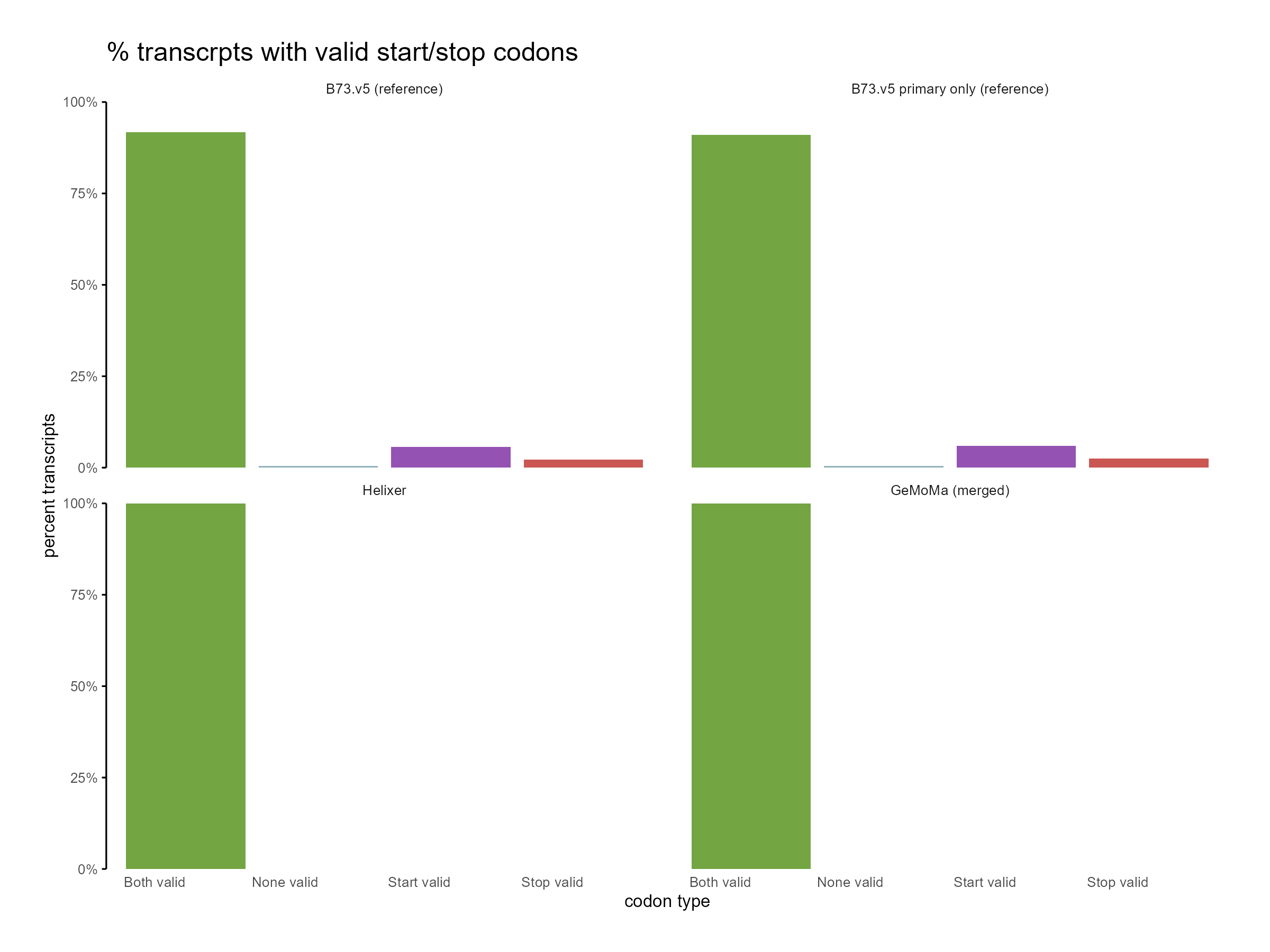
Figure 10: Distribution of start and stop codons for GeMoMa, Helixer and NAM.v5 predictions.